
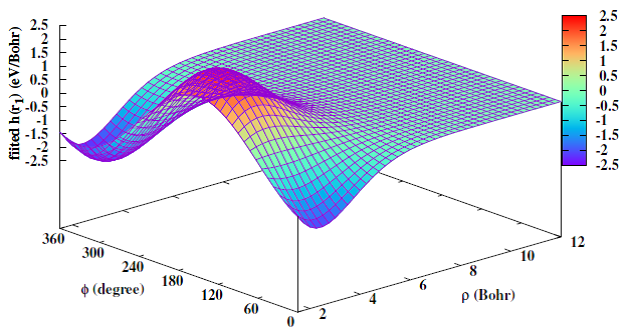
(1)分子体系的精确势能面构造
任何化学过程的所有动力学信息原则上可以先通过构造体系的高精度势能面,再求解原子核运动的薛定谔方程或者牛顿方程得到。因此,精确的势能面构建是分子动力学理论研究的前提和基础。本研究组致力于发展高效精确的基于机器学习的势能面拟合方法,采用尽可能少的从头算能量点,针对重要的分子反应、分子光解和分子团簇等体系和动力学过程,特别是有非绝热效应,比如有锥形交叉的分子体系,构建目前最高精度的全维全域绝热势能面和非绝热势能矩阵,为高精度的动力学模拟奠定基础。

(2)量子态分辨的动力学研究
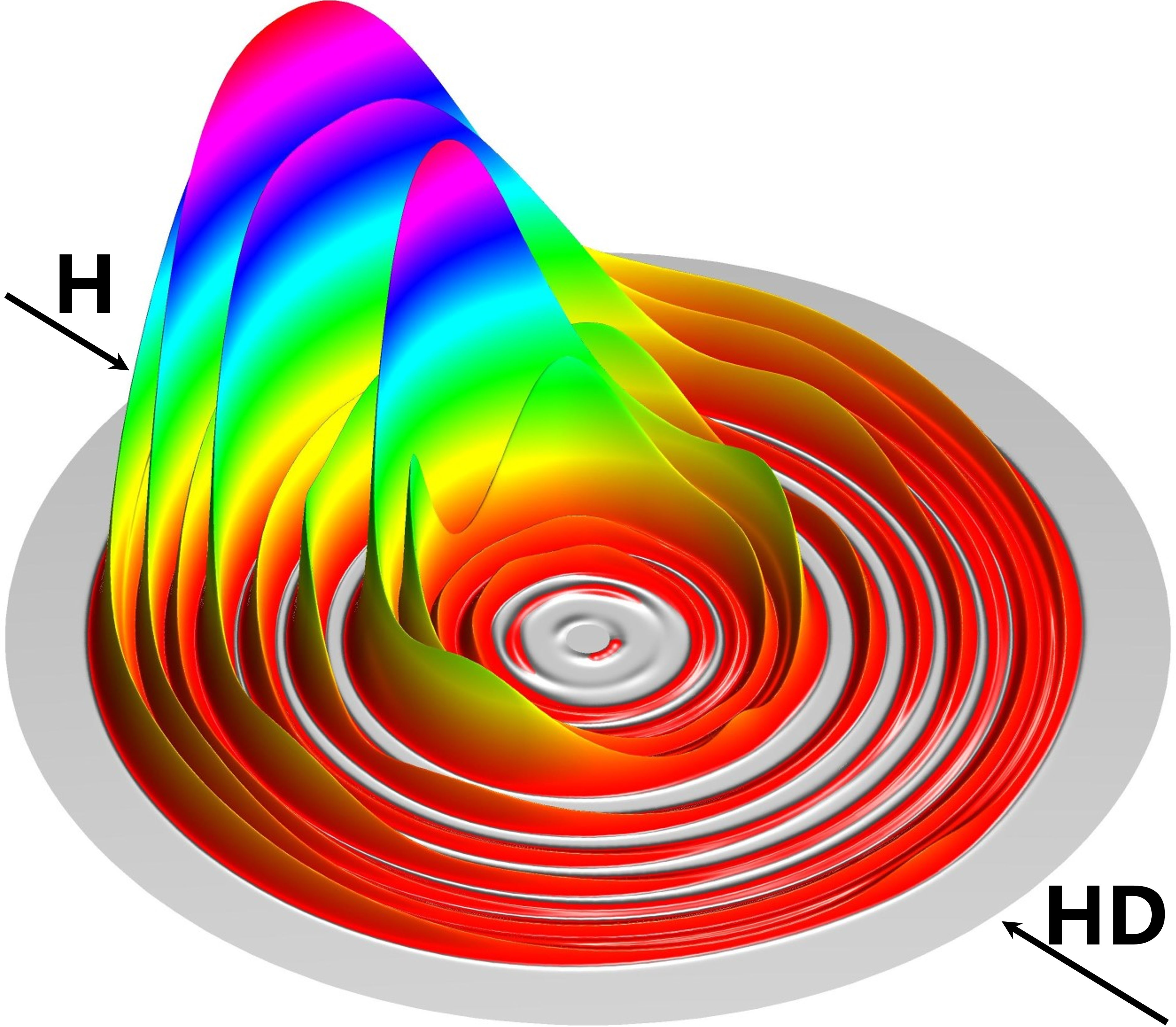
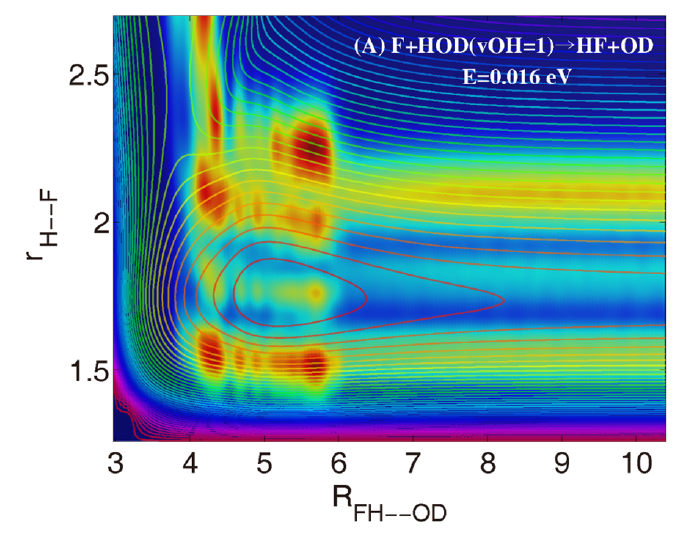
随着体系的增大和自由度的增加,如何发展高精度、高效率的量子动力学理论与计算方法,实现态-态分辨的量子动力学研究,是分子反应动力学理论发展的基础和动力。本研究组致力于发展多原子体系的量子动力学散射方法和高效并行算法,实现复杂多原子体系的高精度量子动力学模拟;发展含时波包动力学方法,研究超低温反应的动力学信息;发展基于RPMD的动力学方法,考虑复杂多原子体系的量子效应。对多原子体系开展最精确的动力学研究,揭示星际化学、燃烧化学、化学激光等重要化学过程中的量子现象和非绝热效应等动力学过程。

(3)分子—表面散射动力学研究
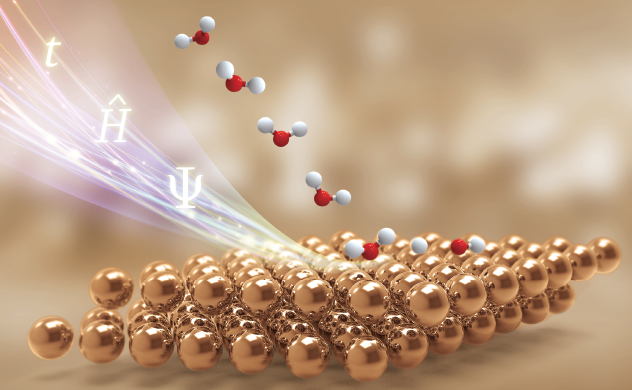
分子在金属表面解离吸附的动力学研究在多相催化等工业过程中占有重要的地位。由于表面的参与,分子在表面的解离吸附问题无论在电子结构计算还是在动力学研究方面都比孤立分子体系研究复杂和困难。本研究组致力于构建目前最为精确的分子解离吸附势能面和发展高维量子动力学方法,对从简单双原子分子到多原子反应在表面的解离体系开展目前最为精确的理论研究。

Copyright © 中国科学院大连化学物理研究所 反应动力学理论与计算研究组 版权所有 All Rights Reserved.